
Aprobaciones de la FDA para 2025
En 2025, la FDA aprobó 46 agentes terapéuticos novedosos de su Center for Drug Evaluation and Research, marcando una ligera disminución respecto al promedio de cinco años, pero por encima de las normas históricas. El cáncer lideró las aprobaciones, con avances en inhibidores de quinasa y terapias génicas. El año introdujo nuevos programas y enfrentó desafíos de personal bajo liderazgo cambiante.
En esta página
- Resumen de las aprobaciones de 2025
- Áreas terapéuticas en foco
- Modalidades emergentes
- Destacados del CBER
- Proyecciones de ventas
- Entorno regulatorio
- Liderazgo en oncología
- Avances no oncológicos
- Otros medicamentos first-in-class
- Avances en enfermedades raras
- Progreso en terapia génica
- Cambios de política y rechazos
- Aprobaciones próximas
Resumen de las aprobaciones de 2025
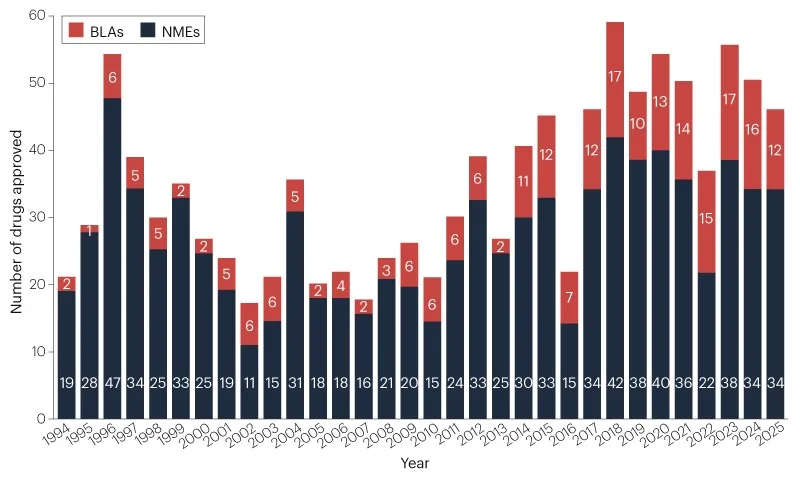
Las compañías farmacéuticas obtuvieron autorización para 46 productos terapéuticos innovadores del Center for Drug Evaluation and Research (CDER) de la FDA durante 2025. Esta cifra reduce ligeramente el promedio móvil de cinco años a 48 nuevos medicamentos anuales, como se ilustra en la Figura 1 y la Tabla 1. No obstante, supera el punto de referencia histórico a largo plazo de 36 aprobaciones por año desde 1993.
Áreas terapéuticas en foco
La oncología sigue siendo el campo predominante para las nuevas autorizaciones de fármacos. De las nuevas aprobaciones del CDER, 16 (35%) se dirigieron a tratamientos contra el cáncer, superando el promedio móvil de cinco años del 29%. La cardiología le siguió con 5 (11%) autorizaciones, y las alergias junto con condiciones inflamatorias representaron 4 (9%) cada una.
Modalidades emergentes
Los desarrolladores están avanzando en una amplia gama de tipos de tratamientos, que incluyen el primer medicamento biológico basado en adnectina, representado en la Figura 3.
Este período también representó el año más grande para inhibidores de quinasa, que constituyeron aproximadamente un tercio de las nuevas aprobaciones de pequeñas moléculas. El remibrutinib (Rhapsido) de Novartis alcanzó el hito de ser el 100.º inhibidor de quinasa en obtener la autorización de la FDA, destacando el crecimiento continuo de esta categoría de fármacos fuera del cuidado del cáncer. "El campo sigue siendo vibrante", señaló Philip Cohen de la Universidad de Dundee en una entrevista con Nature Reviews Drug Discovery.
La FDA aún no ha divulgado la clasificación de estas aprobaciones según categorías regulatorias.
Destacados del CBER
El Center for Biologics Evaluation and Research aprobó 8 elementos nuevos significativos, listados en la Tabla 2, que incluye la primera terapia génica de una entidad sin fines de lucro.
Proyecciones de ventas
Un informe inminente de Boston Consulting Group indica que el ingreso máximo promedio para estas nuevas aprobaciones del CDER y CBER alcanza aproximadamente US$1.2 mil millones. La mediana se sitúa en alrededor de $600 millones.
Entorno regulatorio
Sin embargo, 2025 resultó desafiante para la FDA, caracterizado por cambios significativos en el personal y las políticas tras la designación del presidente Donald Trump de Robert F. Kennedy Jr. para encabezar el Departamento de Salud y Servicios Humanos de EE.UU. Durante los primeros nueve meses, más del 18% del personal del CDER y CBER se fue mediante despidos o renuncias. El CDER experimentó cinco cambios de liderazgo a lo largo del año.
A pesar de estas interrupciones, la organización introdujo nuevas iniciativas y rutas de autorización. Inició el programa piloto del FDA Commissioner's National Priority Voucher (CNPV), diseñado para acortar los períodos de revisión del estándar de 10-12 meses a solo 2 meses. Los críticos, incluidos algunos empleados de la FDA y observadores, argumentan que esta iniciativa podría introducir influencias políticas en las evaluaciones de fármacos y comprometer la integridad regulatoria. Además, la FDA propuso una vía de mecanismo viable para casos en los que los estudios aleatorizados son imprácticos, como tratamientos personalizados n-of-1. También delineó intenciones para eliminar las pruebas de toxicidad en animales.
Se anticipan más modificaciones. Típicamente, la FDA exige dos ensayos clave para aprobaciones completas, pero informes sugieren un cambio hacia requerir solo un ensayo como estándar.
Liderazgo en oncología
El cáncer persiste como el área dominante en el pipeline de desarrollo de la industria. Aunque los medicamentos oncológicos forman la mayoría de las nuevas autorizaciones, contribuyen solo con dos fármacos multibillonarios, cada uno con potencial de ingresos anuales superior a $2 mil millones según proyecciones de Evaluate.
Se espera que la nueva aprobación de mayor ingresos sea la versión subcutánea de pembrolizumab combinada con berahyaluronidase alfa (Keytruda Qlex) de Merck & Co. para múltiples tumores sólidos.
Pembrolizumab, un icónico inhibidor de PD1 aprobado por primera vez en 2014, activa células inmunes contra células cancerosas. Berahyaluronidase alfa, una enzima endoglucosidasa, ayuda en la entrega de anticuerpos al degradar brevemente la matriz extracelular subcutánea para mejorar la permeación y absorción. Aunque la hialuronidasa ha servido como agente dispersor durante años, esta representa la primera autorización para esta forma de proteína modificada. En consecuencia, la formulación de Merck califica como una nueva aprobación.
Se proyecta que pembrolizumab genere alrededor de $32 mil millones en 2025, posicionándolo como el medicamento líder contra el cáncer y el tercero en ventas totales. Los analistas de Evaluate anticipan que la variante subcutánea alcance $9.3 mil millones en su punto máximo.
Akeso Biopharma recibió autorización para su inhibidor de PD1 penpulimab (Penpulimab), aumentando el número de anticuerpos PD1/PDL1 disponibles a 12.
Este año también vio la autorización de dos nuevos conjugados anticuerpo-fármaco (ADCs), que combinan anticuerpos específicos de tejido con agentes citotóxicos potentes. El ADC dirigido a TROP2 datopotamab deruxtecan (Datroway) de Daiichi Sankyo fue aprobado para cáncer de mama hormono-receptor positivo, HER2-negativo después de terapia endocrina y quimioterapia.
Evaluate predice ingresos máximos de $5.4 mil millones para este ADC, contingente a aprobaciones adicionales en una población mayor de cáncer de pulmón de células no pequeñas. El pionero ADC dirigido a TROP2 sacituzumab govitecan (Trodelvy) de Gilead e Immunomedics, inicialmente aprobado de manera acelerada en 2020, está listo para alcanzar $1.4 mil millones en 2025.
La FDA también autorizó el primer ADC dirigido a c-Met de AbbVie, elisotuzumab vedotin (Emrelis). Este anticuerpo portador de inhibidor de microtúbulos está aprobado para NSCLC no escamoso con niveles elevados de proteína c-Met.
El linvoseltamab (Lynozyfic) de Regeneron introduce otro engager de células T bispecífico. Se une a BCMA en células B y CD3 en células T para eliminar células B en el tratamiento del mieloma múltiple. Esto marca el tercer bispecífico BCMA × CD3 disponible y el décimo engager de células T bispecífico en general.
En el ámbito de los inhibidores de quinasa, la FDA emitió una autorización única de combinación dual-novedosa para dos compuestos previamente en investigación. Avutometinib más defactinib (Avmapki Fakzynja Co-Pack) de Verastem combina un inhibidor de MEK con un inhibidor de FAK para abordar cáncer de ovario seroso de bajo grado recurrente con mutaciones KRAS.
Defactinib representa el primer inhibidor de FAK en recibir aprobación. Varios inhibidores de MEK han sido aprobados previamente. La FDA considera estos dos inhibidores de quinasa innovadores como una sola nueva aprobación de fármaco.
"Las personas deberían abrazar más fácilmente las combinaciones novedosas-novedosas", declaró Jonathon Pachter, CSO de Verastem, en Nature Reviews Drug Discovery.
El pionero activador de proteasa caseinolytic mitocondrial P (ClpP) dordaviprone (Modeyso) de Jazz Pharmaceuticals obtuvo aprobación acelerada para gliomas de línea media difusos con mutaciones H3 K27M en pacientes con progresión después de tratamientos previos. ClpP degrada proteínas mitocondriales, y su estimulación se cree que activa la respuesta de estrés integrada, la muerte celular y la liberación de sustancias que contrarrestan los efectos de crecimiento de las mutaciones H3 K27M.
Avances no oncológicos
Las autorizaciones no cancerosas del CDER incluyeron dos prospectos potenciales de megablockbuster con mecanismos novedosos para indicaciones emergentes.
El brensocatib (Brinsupri) de Insmed es el primer inhibidor de DPP1 en el mercado, dirigido a bronquiectasias e ilustrando el potencial de medicamentos enfocados en neutrófilos en condiciones inflamatorias pulmonares.
La bronquiectasia es una enfermedad pulmonar crónica que causa exceso de moco, tos crónica y dilatación de las vías aéreas. Afecta a 350.000 a 500.000 personas en EE.UU., con tratamientos previos limitados a terapia física para remoción de moco y antibióticos para infecciones. La condición se asocia con neutrófilos hiperactivos, células inmunes que secretan péptidos antimicrobianos conocidos como proteasas serina neutrófilas (NSPs) para combatir patógenos. Brensocatib, un inhibidor de DPP1, limita la producción de NSPs, neutralizando estas enzimas inflamatorias sin afectar otras funciones de neutrófilos.
"Esta es una era alentadora: informar a los pacientes sobre opciones próximas que podrían realmente ayudarles", comentó Doreen Addrizzo-Harris, neumonóloga en NYU Langone Health involucrada en ensayos de brensocatib, a Nature Reviews Drug Discovery.
Los analistas proyectan ingresos máximos de $6.3 mil millones para el medicamento, asumiendo extensiones a otros trastornos relacionados con neutrófilos. Sin embargo, en diciembre, la empresa detuvo el desarrollo en rinosinusitis crónica debido a reducción insuficiente en la hinchazón nasal.
El bloqueador del canal de sodio Na1.8 suzetrigine (Journavx) de Vertex ofrece una alternativa vital sin opioides para el alivio del dolor agudo. Desde los 2000, los investigadores han perseguido la inhibición de canales Na tras descubrir que mutaciones en canales Na1.7 causan problemas de dolor persistente, mientras que variantes de pérdida de función bloquean la sensación de dolor. Los inhibidores de Na1.7 han tenido bajo rendimiento clínico, pero Vertex avanzó con el canal Na1.8 análogo.
"Este representa el primer medicamento diseñado exclusivamente para el manejo del dolor", explicó Paul Negulescu, vicepresidente senior de investigación en Vertex, en Nature Reviews Drug Discovery. "Suzetrigine marca el inicio, no el fin, de esta nueva categoría de fármacos."
Los analistas prevén ventas máximas de $3.7 mil millones, impulsadas por la demanda de analgésicos no adictivos y potencial en dolor crónico. Persisten desafíos, ya que la aprobación se basó en no inferioridad a combinaciones acetaminofén-opioide para dolor agudo postquirúrgico, con incertidumbre en otros escenarios agudos. También tiene un costo más alto que opciones genéricas. La efectividad en condiciones crónicas no está probada, con ensayos de Fase III en curso en neuropatía periférica diabética, aunque falló en radiculopatía lumbosacra.
Más adelante en el año, Vertex abandonó el desarrollo del siguiente inhibidor de Na1.8 VX-993 para dolor agudo tras contratiempos en Fase II.
Otros medicamentos first-in-class
Otros agentes pioneros expanden el arsenal farmacológico, aunque con perspectivas de ventas modestas.
Por ejemplo, el gepotidacin (Blujepa) de GSK ofrece una opción nueva y necesaria de antibiótico oral. Similar a los antibióticos quinolónicos desde los 1960, gepotidacin se dirige a topoisomerases tipo IIA bacterianas. Sin embargo, emplea una estructura distinta e interactúa con la enzima en un sitio diferente, ganando su estatus first-in-class. La FDA lo autorizó inicialmente en mayo para infecciones urinarias no complicadas en mujeres y niños de 12 años o más. Para diciembre, obtuvo aprobación para gonorrea.
El zoliflodacin (Nozolvence) de Innoviva Specialty Therapeutics, otro inhibidor único de topoisomerasa tipo II bacteriana, recibió autorización de la FDA para gonorrea. Los estudios de Fase III para zoliflodacin fueron patrocinados y realizados por el Global Antibiotic Research & Development Partnership (GARDP), una organización pública sin fines de lucro que busca disponibilidad global. Innoviva lo adquirió mediante la compra de Entasis Therapeutics, una escisión de AstraZeneca.
Seguimiento preciso para tu cambio
Únete a miles de usuarios que utilizan Shotlee para rastrear con precisión los medicamentos GLP-1 y sus efectos secundarios.
📱 Usar Shotlee Gratis
Únete a miles de usuarios que utilizan Shotlee para rastrear con precisión los medicamentos GLP-1 y sus efectos secundarios.
Los inhibidores de quinasa se están extendiendo a trastornos inflamatorios e inmunes, principalmente mediante compuestos actuantes sobre BTK. Originalmente aprobados para cánceres sanguíneos al eliminar células B malignas, también modulan el comportamiento de células inmunes. Este año, Sanofi obtuvo la primera autorización para rilzabrutinib (Wayrilz) en trombocitopenia inmune, una condición sanguínea inmune. Novartis obtuvo aprobación para remibrutinib (Rhapsido) en urticaria espontánea crónica, o ronchas persistentes.
Los analistas predicen ingresos máximos de $2.1 mil millones para remibrutinib, que está en desarrollo para esclerosis múltiple, miastenia gravis, hidradenitis supurativa y alergias alimentarias. Estiman ventas máximas de $900 millones para el rilzabrutinib de Sanofi.
En contraste, la agencia denegó el inhibidor de BTK tolebrutinib de Sanofi para esclerosis múltiple.
El nerandomilast (Jascayd) de Boehringer Ingelheim surgió como el primer inhibidor de PDE4 para fibrosis pulmonar idiopática (IPF).
Los inhibidores de PDE4 con efectos antiinflamatorios e inmunomoduladores han sido aprobados para condiciones como enfermedad pulmonar obstructiva crónica y psoriasis. Boehringer ahora agrega aprobación para IPF, un trastorno pulmonar progresivo fatal que afecta hasta 3.6 millones globalmente. Nerandomilast selecciona el subtipo PDE4B para reducir la fibrosis e inflamación pulmonar.
Este es el primer nuevo medicamento para IPF en más de una década, siguiendo al nintedanib (Ofev) de Boehringer, un inhibidor multiquinasa aprobado en 2014, y pirfenidona (Esbriet), ahora de Legacy Pharma.
LIB Therapeutics logró la primera aprobación para un tratamiento basado en adnectina con el inhibidor de PCSK9 lerodalcibep (Lerochol).
Los adnectines son biológicos construidos a partir de dominios de fibronectina tipo III, proteínas que apoyan interacciones celulares y pueden unirse a varios ligandos. Durante años, los científicos han propuesto adnectines como alternativas a anticuerpos, ofreciendo targeting preciso para moléculas extracelulares o de superficie celular con diseños compactos y estables en comparación con anticuerpos monoclonales.
LIB utilizó este enfoque para crear el bloqueador de PCSK9 más nuevo, un objetivo validado para reducción de colesterol. Los anticuerpos PCSK9 han estado disponibles por una década, pero su adopción se retrasa por altos costos e inconveniencia de inyección versus píldoras genéricas. LIB anticipa ventajas de lerodalcibep: una dosis subcutánea mensual autoadministrada con estabilidad extendida a temperatura ambiente.
Los inhibidores de PCSK9 de pequeñas moléculas orales podrían llegar pronto. Merck & Co. planea presentar enlicitide, una píldora diaria, para aprobación en abril. Otorgado un CNPV, podría someterse a revisión rápida. Una vez aprobado, la empresa planea ofrecerlo asequiblemente a pacientes de EE.UU.
Avances en enfermedades raras
Las personas con angioedema hereditario (HAE) accedieron a tres nuevos medicamentos.
El HAE es una condición de hinchazón genética derivada de deficiencias o mal funcionamiento del inhibidor C1, un regulador de proteasas serina. Esto lleva a mayor actividad de proteasa del complemento, calicreína plasmática y proteasas de coagulación (factor XIa, XIIa, XIIf).
El anticuerpo profiláctico garadacimab (Andembry) de CSL Behring se dirige al factor XII activado para prevenir episodios. El oligonucleótido antisentido donidalorsen (Dawnzera) de Ionis aborda la prekallicreína para prevenir ataques. El inhibidor de calicreína plasmática de pequeñas moléculas sebetralstat (Ekterly) de Kalvista trata brotes agudos.
Estos entran en un panorama competitivo de HAE con terapias preventivas y agudas existentes que afectan puntos diferentes de la vía.
Los pacientes en riesgo de progresión en nefropatía por inmunoglobulina A primaria (IgAN) recibieron dos tratamientos novedosos. La IgAN, un trastorno renal inmune, involucra acumulación de complejos inmunes IgA causando pérdida gradual de función renal y posible falla. El sibeprenlimab (Voyxact) de Otsuka, un anticuerpo pionero dirigido a APRIL, limita la producción de IgA para reducir proteinuria. El atrasentan (Vanrafia) de Novartis, un bloqueador de receptor de endotelina, reduce la presión glomerular e inflamación renal.
El atacicept de Vera Therapeutics, una proteína de fusión que inhibe BLyS y APRIL, espera decisión de la FDA para IgAN en 2026.
Progreso en terapia génica
El CBER continúa acumulando autorizaciones de terapia génica. Esto abarca la primera aprobación sin fines de lucro, de Fondazione Telethon ETS para etuvetidigene autotemcel (Waskyra) en síndrome de Wiskott-Aldrich (WAS).
El WAS, un trastorno genético por mutaciones en el gen WAS que causan plaquetas defectuosas, se trata con etuvetidigene autotemcel, una terapia génica ex vivo que usa vectores lentivirales para insertar la secuencia WAS en células madre hematopoyéticas del paciente, restaurando la función plaquetaria. Originado en una colaboración de 2010 entre Fondazione Telethon, San Raffaele Telethon Institute for Gene Therapy y GSK, demostró efectividad duradera. Tras luchas de GSK con la comercialización de terapias génicas, se transfirió a Orchard Therapeutics y luego a Fondazione Telethon en 2022.
Fondazione Telethon se asociará con Orphan Therapeutics Accelerator, una biotecnológica sin fines de lucro, para acceso en EE.UU. Anticipan tratar menos de 10 pacientes estadounidenses anuales, con conversaciones de precios en curso. El campo de terapia génica monitorea este marco sin fines de lucro.
"Como investigador, subestimé el esfuerzo en presentar y sostener un fármaco de mercado", compartió Alessandro Aiuti, director adjunto de investigación clínica en San Raffaele Telethon Institute for Gene Therapy, quien ayudó en la aprobación, con Nature Reviews Drug Discovery.
La FDA también autorizó prademagene zamikeracel (Zevaskyn) de Abeona Therapeutics, la primera terapia génica de lámina celular para trastornos cutáneos graves. Involucra células cutáneas del paciente biopsiadas transducidas para expresar colágeno tipo VII, cultivadas en láminas del tamaño de una tarjeta para injertos de heridas. Aprobado para epidermólisis bullosa distrófica recesiva (DEB), una fragilidad cutánea genética por mutaciones en el gen de colágeno tipo VII.
En 2023, el beremagene geperpavec (Vyjuvek) de Krystal Biotech, una terapia génica tópica reutilizable de colágeno tipo VII, fue aprobado para curación de heridas en DEB.
El zopapogene imadenovec (Papzimeos) de Precigen es la primera inmunoterapia para papilomatosis respiratoria recurrente (RRP), un trastorno de crecimiento de vías aéreas causado por VPH. Usa un vector de adenovirus de gorila no replicante con genes VPH 6 y 11 para elicitar respuestas de células T contra VPH, eliminando crecimientos.
El CBER otorgó autorización completa a dos nuevas vacunas contra COVID-19. El mNexspike de Moderna, una vacuna de ARNm avanzada, emplea una quinta parte del ARNm de Spikevax y codifica proteína spike parcial. Los analistas proyectan más de $3.2 mil millones en ventas.
Cambios de política y rechazos
En otro cambio, la FDA comenzó a emitir cartas de respuesta completa parcialmente censuradas para fármacos denegados. Para el 31 de diciembre, listó 43 rechazos en 2025, cubriendo nuevos fármacos (Tabla 3), suplementos, genéricos y biosimilares para CDER o CBER.
Scholar Rock enfrentó una carta de respuesta completa para apitegromab, un anticuerpo anti-promyostatina para atrofia muscular espinal, debido a problemas de fabricación de terceros. Planean resubmisión tras correcciones.
Regeneron recibió un segundo rechazo para odronextamab, un bispecífico CD20 × CD3 para linfoma folicular, en parte por los mismos problemas de fabricación.
Stealth Bio obtuvo un rechazo en mayo para elampretide, un ligador de cardiolipina mitocondrial para síndrome de Barth, por falta de evidencia de eficacia para aprobación completa o acelerada. Resubmitieron con un nuevo punto final y obtuvieron aprobación en septiembre.
La agencia también rechazó vusolimogene oderparepvec de Replimune, potencialmente el primer nuevo virus oncolítico desde talimogene laherparepvec (Imlygic) de Amgen en 2015. Ensayos con nivolumab en melanoma mostraron tasas de respuesta más altas que controles, pero variabilidad poblacional complica la interpretación, y efectos no se separaron de nivolumab. Resubmitieron, con decisión pendiente en abril.
Aprobaciones próximas
Varios medicamentos innovadores podrían recibir autorizaciones iniciales el próximo año (Tabla 4).
El vepdegestrant dirigido al receptor de estrógeno de Arvinas y Pfizer podría ser el primer degradador de proteínas dirigido aprobado. Aunque existen degraders selectivos de receptor de estrógeno, vepdegestrant es un PROTAC de dos brazos que une el receptor a componentes proteasómicos para degradación. Ha luchado por destacarse de otros SERDs, y los socios buscan licenciarlo.
Si se aprueba, afirmaría los degraders dirigidos, potencialmente permitiendo drogar objetivos intratables con variantes de pegamento molecular.
Denali Therapeutics anticipa aprobación para tividenofusp alfa, una terapia de reemplazo enzimático para síndrome de Hunter. El idursulfase (Elaprase) de Takeda, una terapia enzimática IDS desde 2006, no penetra bien el cerebro para problemas neurológicos. Tividenofusp alfa fusiona IDS a un fragmento Fc que se une a receptores de transferrina para entrada al SNC.
Regenxbio podría obtener autorización para clemidsogene lanparvovec, una terapia génica inyectada en SNC para síndrome de Hunter.
Información de la fuente
Publicado originalmente por Nature.Lee el artículo original →