
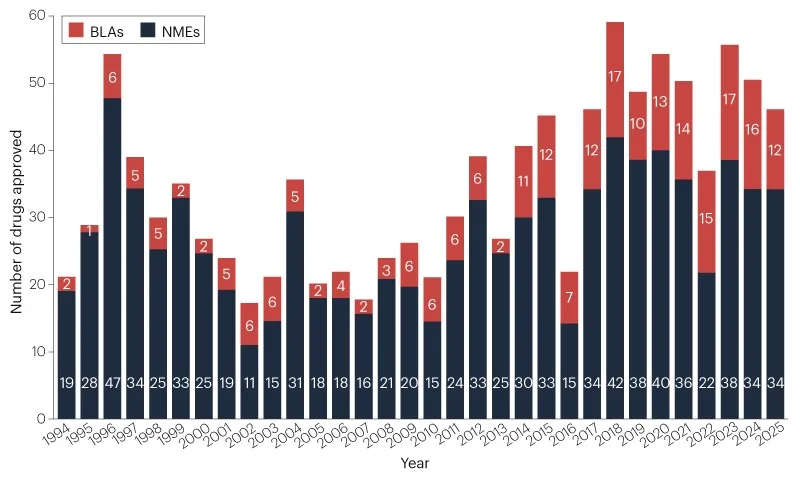
FDA-Zulassungen für 2025
2025 genehmigte die FDA 46 neuartige therapeutische Wirkstoffe aus ihrem Center for Drug Evaluation and Research, was einen leichten Rückgang gegenüber dem Fünf-Jahres-Durchschnitt markiert, aber über historischen Normen liegt. Krebsbehandlungen führten die Zulassungen an, mit Fortschritten bei Kinase-Inhibitoren und Gentherapien. Das Jahr brachte neue Programme und stand vor Personalknappheit unter Führungswechseln.
Auf dieser Seite
- Übersicht über die Zulassungen 2025
- Therapiegebiete im Fokus
- Neue Modalitäten
- CBER-Highlights
- Umsatzprognosen
- Regulatorische Lage
- Onkologie-Führung
- Durchbrüche außerhalb der Onkologie
- Weitere First-in-Class-Arzneimittel
- Fortschritte bei seltenen Erkrankungen
- Fortschritte in der Gentherapie
- Politikänderungen und Ablehnungen
- Kommende Zulassungen
Übersicht über die Zulassungen 2025
Pharmazeutische Unternehmen erhielten 2025 die Zulassung für 46 innovative therapeutische Produkte vom Center for Drug Evaluation and Research (CDER) der FDA. Diese Zahl senkt den gleitenden Fünf-Jahres-Durchschnitt leicht auf 48 neue Arzneimittel pro Jahr, wie in Abbildung 1 und Tabelle 1 dargestellt. Dennoch übertrifft sie den langfristigen historischen Wert von 36 Zulassungen pro Jahr seit 1993.
Therapiegebiete im Fokus
Die Onkologie bleibt das vorherrschende Feld für neue Arzneimittelzulassungen. Von den neuen CDER-Zulassungen richteten sich 16 (35 %) auf Krebsbehandlungen, was den Fünf-Jahres-Durchschnitt von 29 % übersteigt. Die Kardiologie folgte mit 5 (11 %) Zulassungen, und Allergien sowie entzündliche Erkrankungen machten jeweils 4 (9 %).
Neue Modalitäten
Entwickler bringen eine breite Palette von Behandlungsarten voran, einschließlich des ersten adnectin-basierten biologischen Arzneimittels, wie in Abbildung 3 dargestellt.
Dieses Jahr war das größte für Kinase-Inhibitoren, die etwa ein Drittel der neuen Small-Molecule-Zulassungen ausmachten. Novartis’ remibrutinib (Rhapsido) erreichte den Meilenstein als 100. von der FDA zugelassener Kinase-Inhibitor und unterstreicht das anhaltende Wachstum dieser Arzneimittelklasse auch außerhalb der Onkologie. „Das Feld bleibt lebendig“, bemerkte Philip Cohen von der University of Dundee in einem Interview mit Nature Reviews Drug Discovery.
Die FDA hat die Klassifizierung dieser Zulassungen nach regulatorischen Kategorien noch nicht veröffentlicht.
CBER-Highlights
Das Center for Biologics Evaluation and Research genehmigte 8 bedeutende neue Produkte, aufgelistet in Tabelle 2, darunter die erste Gentherapie einer Non-Profit-Organisation.
Umsatzprognosen
Ein bevorstehender Bericht der Boston Consulting Group deutet darauf hin, dass der durchschnittliche Höchstumsatz dieser neuen CDER- und CBER-Zulassungen etwa 1,2 Milliarden US-Dollar beträgt. Der Median liegt bei rund 600 Millionen US-Dollar.
Regulatorische Lage
Allerdings war 2025 eine herausfordernde Zeit für die FDA, geprägt von erheblichen Personal- und Politikwechseln nach der Ernennung von Robert F. Kennedy Jr. durch Präsident Donald Trump zum Leiter des US Department of Health and Human Services. In den ersten neun Monaten schieden über 18 % des CDER- und CBER-Personals durch Entlassungen oder Kündigungen aus. CDER erlebte im Laufe des Jahres fünf Führungswechsel.
Trotz dieser Störungen führte die Behörde neue Zulassungsinitiativen und -wege ein. Sie startete das Pilotprogramm FDA Commissioner’s National Priority Voucher (CNPV), das Prüfzeiten von standardmäßig 10–12 Monaten auf nur 2 Monate verkürzt. Kritiker, darunter FDA-Mitarbeiter und Beobachter, befürchten, dass dieses Programm politische Einflüsse in die Arzneimittelbewertung bringen und die regulatorische Integrität gefährden könnte. Zudem schlug die FDA einen machbaren Mechanismus-Weg für Fälle vor, in denen randomisierte Studien unpraktikabel sind, wie bei personalisierten n-of-1-Behandlungen. Sie skizzierte auch Pläne zur Abschaffung von Tierversuchstoxizitätsprüfungen.
Weitere Änderungen werden erwartet. Normalerweise fordert die FDA für vollständige Zulassungen zwei Pivotalstudien, Berichten zufolge könnte sich der Standard auf eine einzige Studie verschieben.
Onkologie-Führung
Krebs bleibt das dominierende Gebiet in der Entwicklungs-Pipeline der Branche. Obwohl Onkologie-Arzneimittel die Mehrheit der neuen Zulassungen stellen, liefern sie nur zwei Multi-Milliarden-Dollar-Arzneimittel mit einem jährlichen Umsatzpotenzial von über 2 Milliarden US-Dollar nach Evaluate-Prognosen.
Die einträglichste neue Zulassung dürfte die subkutane Version von pembrolizumab in Kombination mit berahyaluronidase alfa von Merck & Co. (Keytruda Qlex) für multiple solide Tumore sein.
Pembrolizumab, ein ikonisches PD1-Inhibitor, das erstmals 2014 zugelassen wurde, aktiviert Immunzellen gegen Krebszellen. Berahyaluronidase alfa, ein Endoglykosidase-Enzym, unterstützt die Antikörperabgabe, indem es die subkutane extrazelluläre Matrix vorübergehend abbaut, um die Permeation und Aufnahme zu verbessern. Hyaluronidase wird seit Jahren als Dispergiermittel eingesetzt, dies ist jedoch die erste Zulassung für diese modifizierte Proteinform. Daher gilt Merck’s Formulierung als neue Zulassung.
Pembrolizumab soll 2025 rund 32 Milliarden US-Dollar einbringen und sich als führendes Krebsarzneimittel – und drittes insgesamt – positionieren. Analysten von Evaluate prognostizieren für die subkutane Variante einen Höchstumsatz von 9,3 Milliarden US-Dollar.
Akeso Biopharma erhielt die Zulassung für seinen PD1-Inhibitor penpulimab (Penpulimab), wodurch die Anzahl verfügbarer PD1/PDL1-Antikörper auf 12 steigt.
In diesem Jahr wurden zwei neue Antibody-Drug-Conjugates (ADCs) zugelassen, die gewebespezifische Antikörper mit starken zytotoxischen Wirkstoffen kombinieren. Datopotamab deruxtecan (Datroway) von Daiichi Sankyo, ein TROP2-gerichtetes ADC, wurde für hormonrezeptor-positive, HER2-negative Brustkrebs nach Endokrin- und Chemotherapie zugelassen.
Evaluate prognostiziert Höchstumsätze von 5,4 Milliarden US-Dollar für dieses ADC, abhängig von weiteren Zulassungen in einer größeren Population mit nicht-kleinzelligem Lungenkarzinom. Sacituzumab govitecan (Trodelvy) von Gilead und Immunomedics, das Pionier-TROP2-ADC mit beschleunigter Zulassung 2020, soll 2025 1,4 Milliarden US-Dollar erreichen.
Die FDA genehmigte auch AbbVies erstes c-Met-gerichtetes ADC elisotuzumab vedotin (Emrelis). Dieses mit Mikrotubuli-Inhibitor beladene Antikörperpräparat ist für nicht-plattenepitheliales NSCLC mit erhöhten c-Met-Proteinspiegeln zugelassen.
Linvoseltamab (Lynozyfic) von Regeneron ist ein weiterer bispezifischer T-Zell-Engager. Es bindet an BCMA auf B-Zellen und CD3 auf T-Zellen, um B-Zellen bei der Behandlung des multiplen Myeloms zu eliminieren. Es ist das dritte verfügbare BCMA × CD3-Bispezifikum und der zehnte bispezifische T-Zell-Engager insgesamt.
Im Bereich Kinase-Inhibitoren erteilte die FDA eine einzigartige Dual-Novel-Kombi-Zulassung für zwei zuvor investigationale Verbindungen. Avutometinib plus defactinib (Avmapki Fakzynja Co-Pack) von Verastem kombiniert einen MEK-Inhibitor mit einem FAK-Inhibitor für KRAS-mutiertes rezidivierendes niedriggradiges seröses Ovarialkarzinom.
Defactinib ist der erste zugelassene FAK-Inhibitor. Mehrere MEK-Inhibitoren wurden zuvor genehmigt. Die FDA betrachtet diese zwei innovativen Kinase-Inhibitoren als eine neue Arzneimittelzulassung.
„Man sollte Novel-Novel-Kombinationen offener umarmen“, sagte Jonathon Pachter, CSO von Verastem, in Nature Reviews Drug Discovery.
Dordaviprone (Modeyso) von Jazz Pharmaceuticals, der erste Aktivator der mitochondrialen Caseinlytic-Protease P (ClpP), erhielt beschleunigte Zulassung für diffuse mittellinig Gliome mit H3 K27M-Mutationen bei Patienten mit Progression nach vorheriger Therapie. ClpP abbaut mitochondriale Proteine, und seine Aktivierung soll die integrierte Stressreaktion, Zelltod und die Freisetzung von Substanzen auslösen, die die wachstumsfördernden Effekte der H3 K27M-Mutationen bekämpfen.
Durchbrüche außerhalb der Onkologie
Zu den nicht-onkologischen CDER-Zulassungen gehörten zwei potenzielle Mega-Blocker mit neuen Wirkmechanismen für aufstrebende Indikationen.
Brensocatib (Brinsupri) von Insmed ist der erste DPP1-Inhibitor auf dem Markt und zielt auf Bronchiektasen ab, was das Potenzial neutrophilenfokussierter Arzneimittel bei Lungenentzündungen zeigt.
Bronchiektasen sind eine chronische Lungenkrankheit mit übermäßigem Schleim, chronischem Husten und Erweiterung der Atemwege. Sie betrifft 350.000 bis 500.000 Personen in den USA, wobei frühere Behandlungen auf Physiotherapie zur Schleimlösung und Antibiotika bei Infektionen beschränkt waren. Die Erkrankung hängt mit hyperaktiven Neutrophilen zusammen, Immunzellen, die antimikrobielle Peptide namens Neutrophilen-Serinhaltige Proteasen (NSPs) zur Bekämpfung von Pathogenen absondern. Brensocatib, ein DPP1-Inhibitor, hemmt die NSP-Produktion und neutralisiert diese entzündlichen Enzyme, ohne andere Neutrophilenfunktionen zu beeinträchtigen.
„Das ist eine ermutigende Zeit – Patienten von bevorstehenden Optionen zu erzählen, die ihnen wirklich helfen könnten“, sagte Doreen Addrizzo-Harris, Pulmonologin am NYU Langone Health und Beteiligte an Brensocatib-Studien, zu Nature Reviews Drug Discovery.
Analysten prognostizieren Höchstumsätze von 6,3 Milliarden US-Dollar, unter der Annahme von Erweiterungen auf andere neutrophilenassoziierte Erkrankungen. Im Dezember stoppte das Unternehmen jedoch die Entwicklung bei chronischer Rhinosinusitis aufgrund unzureichender Reduktion der Nasenschwellung.
Suzetrigine (Journavx) von Vertex, ein Na1.8-Natriumkanalblocker, bietet eine wichtige opioidfreie Alternative zur Akutschmerzbehandlung. Seit den 2000er Jahren verfolgen Forscher die Na-Kanalhemmung, nachdem Mutationen in Na1.7-Kanälen anhaltende Schmerzerkrankungen verursachen und Verlust-of-Function-Varianten Schmerzwahrnehmung blockieren. Na1.7-Inhibitoren scheiterten klinisch, doch Vertex setzte auf den analogen Na1.8-Kanal voran.
„Dies ist das erste Arzneimittel, das ausschließlich für die Schmerzbehandlung entwickelt wurde“, erklärte Paul Negulescu, Senior Vice President Research bei Vertex, in Nature Reviews Drug Discovery. „Suzetrigine ist der Anfang, nicht das Ende dieser neuen Arzneimittelklasse.“
Analysten erwarten Höchstumsätze von 3,7 Milliarden US-Dollar, getrieben durch die Nachfrage nach nicht-suchterzeugenden Analgetika und Potenzial bei chronischen Schmerzen. Herausforderungen bleiben: Die Zulassung basierte auf Non-Inferiorität zu Acetaminophen-Opioid-Kombinationen bei postoperativem Akutschmerz, mit Unsicherheiten bei anderen Akutszenarien. Es ist teurer als Generika. Die Wirksamkeit bei chronischen Zuständen ist unbewiesen, mit laufenden Phase-III-Studien bei diabetischer peripherer Neuropathie, obwohl es bei lumbosakraler Radikulopathie scheiterte.
Später im Jahr gab Vertex die Entwicklung des Nachfolgers Na1.8-Inhibitors VX-993 für Akutschmerz nach Phase-II-Misserfolgen auf.
Weitere First-in-Class-Arzneimittel
Andere Pionierarzneimittel erweitern das pharmakologische Arsenal, wenn auch mit bescheidenen Umsatzprognosen.
GSK’s gepotidacin (Blujepa) bietet eine notwendige neue orale Antibiotikaoption. Ähnlich wie Chinolon-Antibiotika seit den 1960er Jahren zielt gepotidacin auf bakterielle Typ-IIA-Topoisomerasen ab. Es nutzt jedoch einen anderen chemischen Aufbau und bindet am Enzym an einer anderen Stelle, was ihm den First-in-Class-Status verleiht. Die FDA genehmigte es zunächst im Mai für unkomplizierte Harnwegsinfekte bei Frauen und Kindern ab 12 Jahren. Bis Dezember folgte die Zulassung für Gonorrhö.
Zoliflodacin (Nozolvence) von Innoviva Specialty Therapeutics, ein weiterer einzigartiger bakterieller Typ-II-Topoisomerase-Inhibitor, erhielt FDA-Zulassung für Gonorrhö. Phase-III-Studien für zoliflodacin wurden von der Global Antibiotic Research & Development Partnership (GARDP), einer öffentlichen Non-Profit-Organisation für globale Verfügbarkeit, gesponsert und durchgeführt. Innoviva erwarb es durch den Kauf von Entasis Therapeutics, einem AstraZeneca-Ableger.
Präzises Tracking auf Deinem Weg
Begleite Tausende Nutzer, die mit Shotlee ihre GLP-1-Medikamente und Nebenwirkungen exakt tracken.
📱 Shotlee kostenlos nutzen
Begleite Tausende Nutzer, die mit Shotlee ihre GLP-1-Medikamente und Nebenwirkungen exakt tracken.
Kinase-Inhibitoren dringen in entzündliche und Immunerkrankungen vor, hauptsächlich über BTK-wirkende Verbindungen. Ursprünglich für Blutkrebs zugelassen, indem sie maligne B-Zellen eliminieren, modulieren sie auch Immunzellverhalten. Sanofi sicherte die erste Zulassung für rilzabrutinib (Wayrilz) bei immunthrombozytopenischer Purpura, einer immunbedingten Bluterkrankung. Novartis erhielt die Zulassung für remibrutinib (Rhapsido) bei chronischer spontaner Urtikaria, also persistierenden Nesselsucht.
Analysten prognostizieren Höchstumsätze von 2,1 Milliarden US-Dollar für remibrutinib, das für Multiple Sklerose, Myasthenia gravis, Hidradenitis suppurativa und Nahrungsmittelallergien entwickelt wird. Für Sanofis rilzabrutinib schätzen sie 900 Millionen US-Dollar.
Im Gegensatz dazu lehnte die Behörde Sanofis BTK-Inhibitor tolebrutinib für Multiple Sklerose ab.
Nerandomilast (Jascayd) von Boehringer Ingelheim ist der erste PDE4-Inhibitor für idiopathische Lungenfibrose (IPF).
PDE4-Inhibitoren mit entzündungshemmenden und immunmodulatorischen Effekten sind für Erkrankungen wie chronisch obstruktive Lungenerkrankung und Psoriasis zugelassen. Boehringer ergänzt nun IPF, eine progressive tödliche Lungenkrankheit, die bis zu 3,6 Millionen Menschen weltweit betrifft. Nerandomilast zielt selektiv auf den PDE4B-Subtyp ab, um Fibrose und Lungenentzündung zu reduzieren.
Es ist das erste neue Arzneimittel für IPF seit über einem Jahrzehnt, nach nintedanib (Ofev) von Boehringer, einem Multikinase-Inhibitor aus 2014, und pirfenidone (Esbriet), nun von Legacy Pharma.
LIB Therapeutics erzielte die erste Zulassung für ein adnectin-basiertes Therapeutikum mit dem PCSK9-Inhibitor lerodalcibep (Lerochol).
Adnectine sind Biologika aus Fibronectin-Typ-III-Domänen, Proteinen, die Zellinteraktionen unterstützen und an verschiedene Liganden binden können. Seit Jahren werden Adnectine als Antikörperalternativen vorgeschlagen, mit präziser Zielrichtung auf extrazelluläre oder zelloberflächenständige Moleküle durch kompakte, stabile Strukturen im Vergleich zu monoklonalen Antikörpern.
LIB nutzte diesen Ansatz für den neuesten PCSK9-Blocker, ein validiertes Ziel zur Cholesterinsenkung. PCSK9-Antikörper gibt es seit einem Jahrzehnt, doch die Akzeptanz hinkt wegen hoher Kosten und Injektionsbelastung hinter generischen Pillen her. LIB erwartet Vorteile für lerodalcibep – monatliche subkutane Selbstinjektion mit hoher Stabilität bei Raumtemperatur.
Orale Small-Molecule-PCSK9-Inhibitoren könnten bald kommen. Merck & Co. plant die Einreichung von enlicitide, einer täglichen Pille, im April. Mit einem CNPV könnte es schnell geprüft werden. Bei Zulassung will das Unternehmen es erschwinglich für US-Patienten anbieten.
Fortschritte bei seltenen Erkrankungen
Patienten mit hereditärem Angioödem (HAE) erhielten Zugang zu drei neuen Arzneimitteln.
HAE ist eine genetische Schwellenerkrankung durch Defizienz oder Funktionsstörung des C1-Inhibitors, eines Serinprotease-Regulators. Dies führt zu erhöhter Aktivität von Komplementproteasen, Plasma-Kallikrein und Gerinnungsproteasen (Faktor XIa, XIIa, XIIf).
Garadacimab (Andembry), der prophylaktische Antikörper von CSL Behring, zielt auf aktivierten Faktor XII ab, um Anfälle zu verhindern. Donidalorsen (Dawnzera), die Antisense-Oligonukleotid von Ionis, adressiert Präkallikrein zur Attacke-Prävention. Sebetralstat (Ekterly), der Small-Molecule-Plasma-Kallikrein-Inhibitor von Kalvista, behandelt akute Schübe.
Diese treten in ein wettbewerbsintensives HAE-Umfeld mit bestehenden präventiven und akuten Therapien ein, die unterschiedliche Wegpunkte beeinflussen.
Patienten mit Risiko für Progression bei primärer Immunglobulin-A-Nephropathie (IgAN) erhielten zwei neue Behandlungen. IgAN, eine immune Nierenerkrankung, beinhaltet IgA-Immunkomplex-Ablagerungen, die zu schrittweisem Verlust der Nierenfunktion und potenzieller Versagen führen. Sibeprenlimab (Voyxact) von Otsuka, ein Pionier-APRIL-gerichteter Antikörper, hemmt die IgA-Produktion, um Proteinurie zu mindern. Atrasentan (Vanrafia) von Novartis, ein Endothelin-Rezeptor-Blocker, reduziert glomerulären Druck und Nierenentzündung.
Atacicept von Vera Therapeutics, ein Fusionsprotein, das BLyS und APRIL hemmt, wartet auf FDA-Entscheidung für IgAN 2026.
Fortschritte in der Gentherapie
CBER häuft Gentherapie-Zulassungen an. Dazu gehört die erste Non-Profit-Zulassung für etuvetidigene autotemcel (Waskyra) von Fondazione Telethon ETS bei Wiskott-Aldrich-Syndrom (WAS).
WAS, eine genetische Störung durch WAS-Genmutationen mit defekten Thrombozyten, wird mit etuvetidigene autotemcel behandelt, einer ex-vivo-Gentherapie mit lentiviralen Vektoren, die die WAS-Sequenz in patienteneigene hämatopoetische Stammzellen einbaut und Thrombozytenfunktion wiederherstellt. Aus einer 2010er-Kollaboration von Fondazione Telethon, San Raffaele Telethon Institute for Gene Therapy und GSK entstanden, zeigte es langanhaltende Wirksamkeit. Nach GSKs Problemen mit Gentherapie-Kommerzialisierung ging es an Orchard Therapeutics, dann 2022 an Fondazione Telethon.
Fondazione Telethon kooperiert mit Orphan Therapeutics Accelerator, einer Non-Profit-Biotech, für US-Zugang. Sie erwarten <10 US-Patienten jährlich, mit laufenden Preisprüfungen. Das Gentherapie-Feld beobachtet dieses Non-Profit-Modell.
„Als Forscher habe ich die Anstrengungen für Einreichung und Markterhaltung eines Arzneimittels unterschätzt“, teilte Alessandro Aiuti, stellvertretender Direktor klinischer Forschung am San Raffaele Telethon Institute for Gene Therapy, das die Zulassung unterstützte, mit Nature Reviews Drug Discovery.
Die FDA genehmigte prademagene zamikeracel (Zevaskyn) von Abeona Therapeutics, die erste Zellblatt-Gentherapie für schwere Hauterkrankungen. Sie verwendet biopsierte Patientenhautzellen, transduziert zur Typ-VII-Kollagen-Expression, zu kartengrößen Blättern gezüchtet für Wundtransplantate. Zugelassen für rezessive dystrophische Epidermolysis bullosa (DEB), eine genetische Hautzerbrechlichkeit durch Typ-VII-Kollagen-Genmutationen.
2023 wurde beremagene geperpavec (Vyjuvek) von Krystal Biotech, eine topische wiederholbar anwendbare Typ-VII-Kollagen-Gentherapie, für DEB-Wundheilung zugelassen.
Zopapogene imadenovec (Papzimeos) von Precigen ist die erste Immuntherapie für rezidivierende respiratorische Papillomatose (RRP), eine HPV-bedingte Atemwegs-Wucherung. Es nutzt einen nicht-replizierenden Gorilla-Adenovirus-Vektor mit HPV-6- und -11-Genen, um T-Zell-Antworten gegen HPV zu erzeugen und Wucherungen zu beseitigen.
CBER erteilte vollständige Zulassungen für zwei neue COVID-19-Impfstoffe. Modernas mNexspike, ein fortschrittlicher mRNA-Impfstoff, verwendet ein Fünftel der mRNA von Spikevax und kodiert für partielles Spike-Protein. Analysten prognostizieren über 3,2 Milliarden US-Dollar Umsatz.
Politikänderungen und Ablehnungen
In einer weiteren Veränderung begann die FDA, teilweise zensierte Complete Response Letters für abgelehnte Arzneimittel zu veröffentlichen. Bis 31. Dezember listete sie 43 Ablehnungen 2025 auf, einschließlich neuer Arzneimittel (Tabelle 3), Supplements, Generika und Biosimilars für CDER oder CBER.
Scholar Rock erhielt einen Complete Response Letter für apitegromab, einen Anti-Promostatin-Antikörper für spinale Muskelatrophie, wegen Drittanbieter-Fertigungsmängel. Sie planen Wiedereinreichung nach Behebung.
Regeneron bekam eine zweite Ablehnung für odronextamab, ein CD20 × CD3-Bispezifikum für follikuläres Lymphom, teilweise wegen gleicher Fertigungsprobleme.
Stealth Bio erhielt im Mai eine Ablehnung für elampretide, einen mitochondrialen Cardiolipin-Binder für Barth-Syndrom, mangels Wirksamkeitsnachweis für volle oder beschleunigte Zulassung. Sie reichten mit neuem Endpunkt nach und erhielten im September Zulassung.
Die Behörde wies auch vusolimogene oderparepvec von Replimune ab, potenziell das erste neue onkolytische Virus seit talimogene laherparepvec (Imlygic) von Amgen 2015. Studien mit nivolumab bei Melanom zeigten höhere Response-Raten als Kontrollen, doch Populationsvariabilität erschwert Interpretation, und Effekte waren nicht von nivolumab getrennt. Sie reichten nach, Entscheidung ausstehend im April.
Kommende Zulassungen
Mehrere innovative Arzneimittel könnten nächstes Jahr erste Zulassungen erhalten (Tabelle 4).
Vepdegestrant von Arvinas und Pfizer, ein östrogenrezeptor-gerichteter Degrader, könnte der erste Protein-Degrader-Zulassung sein. Selektive Östrogenrezeptor-Degrader existieren, vepdegestrant ist ein zweigeteilter PROTAC, der das Rezeptor an Proteasom-Komponenten bindet. Es kämpft um Abgrenzung von anderen SERDs, Partner suchen Lizenznehmer.
Bei Zulassung würde es Targeted Degrader bestätigen und potenziell unzugängliche Ziele mit Molecular-Glue-Varianten ermöglichen.
Denali Therapeutics erwartet Zulassung für tividenofusp alfa, ein Enzymersatz für Hunter-Syndrom. Idursulfase (Elaprase) von Takeda, IDS-Enzymtherapie seit 2006, dringt schlecht ins Gehirn für neurologische Symptome vor. Tividenofusp alfa fusioniert IDS mit Fc-Fragment, das Transferrin-Rezeptoren bindet, für ZNS-Eintritt.
Regenxbio könnte Zulassung für clemidsogene lanparvovec, eine CNS-injizierte Gentherapie für Hunter-Syndrom, erhalten.
Quellenangabe
Ursprünglich veröffentlicht von Nature.Originalartikel lesen →